

合作交流
2018必須關注的31個單抗藥物
來源:藥事縱橫
|
作者:派金生物
|
發布時間: 2018-01-14
|
23255 次瀏覽
|
分享到:
概述
單抗即單克隆抗體(Monoclonal antibody,Mab),自1986年第一個單抗產品問世以來,全球已近有80個單抗上市。截止目前單抗已經發展到了四代,第一代為鼠源單抗(momab),第二代為人鼠嵌合型單抗(ximab),第三代為人源化單抗(zumab),第四代為全人源化單抗(mumab)。人源化單抗的優勢在于可以克服人抗鼠抗體反應,可避免單抗分子被免疫系統當作異源蛋白而被快速清除,提高單抗分子的生物學活性。

(圖片來自網絡)
盡管產品不多,然而尚在市場活躍的四十余個單抗卻撐起全球近1000億美金的市場,而且單抗的用藥市場增長速度遠高于同行業的其它水平。優良的療效,龐大的市場以及超高的附加值,催生出大量開發單抗產品的公司,近年來全球單抗行業的發展速度已經顯著加快。2017年美國和歐洲首次獲批的單抗首次達到2位數水平,2018年上市的單抗有望進一步達到新高。2017年12月1日,已經有9個單抗產品處在FDA/EMA的審評之中,有12個產品已經拿到或即將拿到關鍵性臨床的數據,它們有望在2018年內提交上市申請。除此之外,還有近20個單抗產品已經處在臨床末期階段,在不遠的未來,必定是單抗產品“百舸爭流”的時代。
2017歐美批準的單抗回顧
截止2017年12月1日,有10個單抗藥物在美國和歐洲獲得首次批準,除brodalumab已經于2016 年獲得日本批準和sarilumab于2017年1月獲得加拿大批準外,其它產品都是全球首批。這是10個單抗藥物,有5個屬于免疫領域,4個是抗腫瘤領域,1個是血液病領域。

2017歐美批準的單抗回顧
截止2017年12月1日,有10個單抗藥物在美國和歐洲獲得首次批準,除brodalumab已經于2016 年獲得日本批準和sarilumab于2017年1月獲得加拿大批準外,其它產品都是全球首批。這是10個單抗藥物,有5個屬于免疫領域,4個是抗腫瘤領域,1個是血液病領域。
Brodalumab
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827)是一種人IgG2抗體,靶點是白介素-17受體(IL-17RA),可通過IL-17RA阻斷IL-17A的炎癥信號,促炎性細胞因子IL-17F和IL-17C。Brodalumab最早于2016年7 月4日在日本首次獲批,商品名為Lumicef,獲批適應癥為紅皮病型銀屑病、膿皰性銀屑病、銀屑病關節炎和尋常性銀屑病。2017年2月和2017年7月,Brodalumab分別又獲得美國和歐洲批準,用于對系統性療法或光照療法(紫外線治療)不響應的成人中重度斑塊狀銀屑病的治療。Brodalumab的安全有效性在3項安慰劑對照的三期臨床試驗中進行了研究,AMAGINE-1(NCT01708590),AMAGINE-2(NCT01708603)和AMAGINE-3(NCT01708603)三項試驗共納入患者4373名。主要終點是第12周銀屑病面積與嚴重程度改善達75%以上(PASI75)和醫生靜態總體評估(sPGA)得分(0/1)比基線至少下降2分的患者比例。AMAGINE-1結果顯示,Brodalumab 210mg 達PASI75的比例為83%,140mg組為60%,而安慰劑組為3%。sPGA得分達標率同樣高于安慰劑組,分別為76%,54%和1%。AMAGINE-2和AMAGINE-3試驗數據顯示,本品對中重度銀屑病臨床癥狀改善情況明顯優于安慰劑組。AMAGINE-2中210mg 組PASI達標率為86%,140mg組為67%,而安慰劑組為8%,AMAGINE-3則依次為86%,69%和6%,本品治療組sPGA達標率同樣更高,AMAGINE-2中210mg組達標率為79%,140mg組為58%,安慰劑組為4%; AMAGINE-3中,則依次為80%,60%和4%。
Avelumab
Avelumab (Bavencio,MSB0010718C)是一種以PD-L1為靶點的人IgG1單抗,首次批準于2017年5月23日。獲批適應癥為成人和12歲以上兒童的轉移性默克爾細胞癌。2017年9月,該適應癥也在歐洲獲批。Avelumab的獲批是基于二期關鍵性臨床試驗Javelin Merkel 200研究(NCT02155647) 的數據,該試驗包括兩部分,Part A患者事先經過化療,Part B患者則使用Avelumab初次治療。在10mg/kg的劑量下,每兩周一次靜注。Part A中,88名事先經過化療的患者使用本品治療后病情得到了控制,中位隨訪期為10.4個月,客觀緩解率為31.8%,其中8%完全緩解,20%部分緩解。Part B中,39名使用本品首次治療的患者,主要終點中位緩解時間的數據有望在2019年9月獲得。此前,Avelumab在歐洲和澳大利亞均獲得了默克爾細胞癌的孤兒藥地位,還獲得FDA突破性療法認定。2017年5月9日,FDA授予avelumab用于經鉑化療后進展的局部晚期或轉移性尿路上皮癌適應癥優先審評。除此以外,本品用于非小細胞肺癌、腎細胞癌、卵巢癌和胃癌治療的4項三期臨床試驗將在2018年達到終點,而且被美國和歐洲雙雙授予胃癌的孤兒藥資格。
Dupilumab
Dupilumab (Dupixent,REGN668/SAR231893)是一種以IL4R為靶點的IgG4單抗,于2017年4月28日和2017年9月28日分別獲得FDA和EMA批準用于特應性皮炎治療。Dupixent的獲批是基于一項名為LIBERTY的特應性皮炎研究項目,該項目包括SOLO1,SOLO2和CHRONOS三項臨床試驗。在SOLO1和SOLO2中,患者分別每周一次接受300mg Dupixent或安慰劑治療,或每兩周一次接受300mg Dupixent治療后,換用安慰劑治療。CHRONOS試驗則是以安慰劑為對照研究Dupixent與皮質類固醇聯合用藥的療效。試驗的主要終點為16周時研究者整體評價法(IGA)得分(0/1)比基線下降大于2分的患者比例。結果顯示,Dupixent單藥治療16周,IGA達標情況顯著高于安慰劑組,SOLO1為37%,SOLO2為36%。CHRONOS的試驗結果與此相似,dupilumab+皮質類固醇聯合治療組為39%,而安慰劑組+類固醇組為12%。因為療效卓著,本品獲得FDA的特應性皮炎的突破性療法認定。除特應性皮炎之外,dupilumab還在開展哮喘和鼻息肉方面的適應癥,目前處在三期臨床階段。2017年9月,賽諾菲/再生元宣布dupilumab用于無法控制的持續性哮喘的LIBERTY ASTHMA QUEST (NCT02414854)試驗達到治療終點。另外兩項關于鼻息肉治療的臨床試驗(NCT02912468, NCT02898454)也將在2018年達到終點。更可喜的是本品還能用于嗜酸性食道炎治療,目前研究處于臨床二期階段,而且還獲得了FDA的孤兒藥認定。
Ocrelizumab
Ocrelizumab (Ocrevus)是一種人源化的IgG1單抗,作用靶點是CD-20陽性的B細胞。這種B細胞可通過產生促炎性細胞因子、分泌自身抗體和活化促炎性T細胞,而在髓鞘損傷和多發性硬化癥發病中發揮作用。Ocrevus主要通過三種方式消滅CD20陽性B細胞:補體依賴的細胞毒作用和抗體依賴的細胞介導的細胞毒作用,以及在靶細胞上與CD20結合而使其凋亡。這種單抗在內風濕性關節炎(RA)和系統性紅斑狼瘡(SLE)上都進行了臨床研究,但預期效果不理想。Ocrevus于3月29日獲得FDA批準用于復發緩解型的多發性硬化以及原發進展型多發性硬化治療,此前該適應癥獲得了FDA突破性療法認定、快速通道和優先審評資格。OPERAI(NCT01247324)和OPERAII(NCT01412333)兩項試驗對Ocrevus治療發緩解型的多發性硬化的安全和有效性分別進行了評估,結果顯示Ocrevus治療組的年華復發率顯著低于β干擾素組,其中OPERAI低46%,而OPERAII低57%。原發進展型多發性硬化方面,ORATORIO試驗的結果顯示,與安慰劑相比,Ocrevus使12周(主要終點)的臨床殘疾進展顯著降低(32.9% vs 39.3%)。另外MRI顯示,接受Ocrevus治療的患者腦病變的總體積下降3.4%,而安慰劑增加7.4%。
Durvalumab
Durvalumab (Imfinzi,MEDI4736)是一種人IgG1抗體,可直接激活PD-L1以阻斷PD-L1及其受體(PD-1, CD80)間的聯系。Durvalumab設計初衷是阻斷細胞毒性效應與PD-L1陽性免疫細胞間的作用,于2017年5月1日獲FDA加速批準用于鉑化療惡化的局部晚期或轉移性尿路上皮癌。這是一個集優先審評和突破性療法于一身的產品,此次獲批是基于一項182名患者參與的1/2期臨床試驗(NCT01693562)。以10mg/kg的劑量每二周一次靜脈注射Durvalumab,連續用藥12個月,PD-L1高表達亞組的客觀緩解率為27.4%,PD-L1低表達亞組為4.1%。此外,鉑化療進展后局部晚期或無法手術治療的非小細胞肺癌(NSCLC)適應癥NDA已進入FDA審評階段,此前該申請獲得了FDA優先審評和突破性療法認定。與此同時,該適應癥的上市許可申請(MAA)也獲得EMA受理,三期PACIFI臨床試驗(NCT02125461)的數據也已一同提交到EMA。臨床結果顯示化療后進展的NSCLC,以化療為背景添加本品治療,中位無進展生存期為16.8月,而安慰劑只有5.6月。除以上兩個適應癥外,Durvalumab治療PD-L1陽性的轉移性頭頸癌還被FDA授予快速通道資格,三期臨床試驗EAGLE(NCT02369874)和KESTREL(NCT02551159)有望在2018年2月和3月分別達到終點。
Sarilumab
Sarilumab (Kevzara,SAR153191,REGN88)是一種以IL-6R為靶點的IgG1單抗,2017年2月在加拿大首次獲批,2017年5月和2017年7月分別又獲FDA和EMA批準用于對一種或多種生物制品或非生物制品疾病調節性抗風濕藥物(DMARDs)不充分響應或不耐受的中重度活動性類風濕性關節炎。Sarilumab的獲批是基于一項全球多中心的SARIL-RA臨床研究項目,其包括MOBILITY (NCT01061736),TARGET (NCT01709578)和MONARCH (NCT02332590) 三項三期臨床試驗。在MOBILITY和TARGET試驗中,sarilumab均與甲氨蝶呤或其他DMARD合用。MOBILITY研究結果顯示,200mg sarilumab治療組24周ACR20達66%,150mg組為58%,而安慰劑組僅33%。Sarilumab同樣可改善患者生理功能,200mg sarilumab治療組HAQ-DI評分下降0.58,150mg組為0.54,安慰劑組為0.30。TARGET試驗結果與前者相似,DMARD+200mg sarilumab治療組 ACR20達61%,150mg組為56%,而安慰劑組為34%。生理功能改善方面,DMARD+200mg sarilumab治療組HAQ-DI評分下降0.50,150mg組下降0.49,而安慰劑組僅0.29。MONARCH試驗則以阿達木單抗為對照評估sarilumab的安全有效性。24周治療結果顯示,使用紅細胞沉降率計算的28個關節的疾病活動度評分(DAS28-ESR)從基線下降3.28,而阿達木單抗組則只下降2.20,該試驗也達到主要終點。
Guselkumab
Guselkumab (Tremfya) 是一種人IgG1λ單抗,能夠阻滯IL-23的p19亞基,從而限制炎癥響應。IL-23是一種參與Th17細胞分化和維持的細胞因子,通過T細胞亞群生成促炎性細胞因子IL-17、IL-22和TNF。2017年7月13日,guselkumab獲得FDA批準用于適合系統療法(注射或口服治療)或光治療(紫外線治療)的中重度斑塊型銀屑病治療,2017年11月,guselkumab再次獲得EMA批準,適應癥與美國相同。Guselkumab獲批是基于三項臨床試驗的結果,分別為VOYAGE1(NCT02207231),VOYAGE2(NCT02207244)和NAVIGATE(NCT02203032)。其中VOYAGE1和VOYAGE2評估了Tremfya相對于安慰劑和阿達木單抗的療效和安全性,結果顯示,guselkumab治療16周,IGA達標率遠高于安慰劑組(85.1% vs 6.9%),另外,Tremfya治療組分別有73.3%和70.0%的患者實現PASI90 緩解,而阿達木單抗治療組分別為49.7%和46.8%。NAVIGATE試驗則評估了Tremfya相對于Stelara的療效和安全性,患者在0、4周接受ustekinumab治療,16周后響應不足的患者分別再次給予ustekinumab或guselkumab治療,主要終點是28周的IGA評分相比基線下降最低2分的患者比例。而結果顯示經ustekinumab或guselkumab治療,28周IGA評分達標率分別為14.3%和31.1%,32周時則分別為17.3%和36.3%,數據說明Tremfya在既往接受Stelara治療應答不足的患者中仍具有顯著的療效。
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa,CMC-544)是一種抗CD22的人源化IgG4抗體與細胞毒類藥物卡奇霉素的偶聯物。Besponsa于2017年6月30日在歐洲首次獲批,2017年8月17日再次獲得FDA批準,適應癥均為成人復發性或難治性B細胞前體急性淋巴細胞白血病(ALL)。在此此前,Besponsa已經獲得歐美雙方的孤兒藥資格,和FDA的ALL突破性療法認定。Besponsa的獲批是基于一項名為INOVATE ALL三期臨床試驗(NCT01564784),該試驗以現有的標準療法進行對照。患者在第1天(0.8mg/m2)、第8天(0.5mg/m2)和第15天(0.5mg/m2)分別接受三個劑量的Besponsa治療或標準化療。起始療程為21天,此后每個療程均為28天,直達6個療程。結果顯示Besponsa組患者完全緩解率為80.7%,中位總生存期為7.7個月,標準化療組完全緩解率為29.4%,中位總生存期為6.7個月。
Benralizumab
Benralizumab (Fasenra,MEDI-563) 是一個去巖藻糖基化的IgG1單抗,靶點為IL-5R的α亞基。 FDA于2017年11月14日批準本品用于12 歲以上12歲及以上具有嗜酸性表型的重度哮喘 患者的附加維持治療,2017年11月10日,EMA的專家委員會也對本品的上市給出積極的意見。本品的安全有效性WINDWARD研究項目中得到評估,該項目包括SIROCCO,CALIMA,ZONDA,BISE,BORA和GREGALE等6項三期臨床試驗。其中SIROCCO(NCT01928771)和CALIMA (NCT01914757)兩項隨機、雙盲、平行、安慰劑控制的臨床試驗評價了本品長期用藥安全性,患者長期以30mg的固定劑量皮下注射benralizumab長達56周,研究的結果在2016年公開。在為期28周的ZONDA(NCT02075255)試驗中,中重度哮喘患者在口服糖皮質激素的基礎上每四周或8周一次注射Benralizumab 30mg 或安慰劑對病情維持治療,結果顯示,Benralizumab組糖皮質激素用量降低75%,而安慰劑組僅下降 25%。
Emicizumab
Emicizumab (Hemlibra,emicizumab-kxwh,ACE910,RO5534262) 是一種雙特異性IgG4抗體,靶點為IXa和X,2017年11月16日首次獲得FDA批準用于預防或減少體內含有凝血因子VIII抑制物的A型血友病患者的出血頻率。在此之前,Emicizumab已經獲得FDA的孤兒藥和突破性療法認定。日本和歐洲的上市許可申請審評中,歐洲被授予加速審評資格,日本則給予孤兒藥認定。Emicizumab最早由中外發現,與羅氏一同開發并推向全球推向市場。本品的獲批是基于HAVEN1(NCT02622321)的臨床研究結果和HAVEN2(NCT02795767)的中期結果。HAVEN1研究表明,12歲及以上的體內含有VIII因子抑制物的A型血友病患者在接受Hemlibra預防治療后,與沒有接受預防患者相比,年出血率顯著降低87%(2.9 vs 23.3)。AVEN2研究的中期結果表明,12歲以下的體內含有抑制物的A型血友病兒童患者,在接受Hemlibra預防后,有87%未出現出血。在參加NIS 的13名兒童患者的患者內分析中,Hemlibra預防治療與接受旁路制劑治療(BPA)的患者相比出血率降低99%。
處于歐美審評中的單抗
截止2017年12月1日,一共有9個單抗產品在歐美的審評中,除 mogamulizumab于2012年已經在日本獲批外,ibalizumab,burosumab,tildrakizumab,caplacizumab,erenumab,fremanezumab,galcanezumab,romosozumab 尚未在任何國家批準。這些產品有望在2018年問世,其中burosumab和ibalizumab有望在1月或2月獲批上市。
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827)是一種人IgG2抗體,靶點是白介素-17受體(IL-17RA),可通過IL-17RA阻斷IL-17A的炎癥信號,促炎性細胞因子IL-17F和IL-17C。Brodalumab最早于2016年7 月4日在日本首次獲批,商品名為Lumicef,獲批適應癥為紅皮病型銀屑病、膿皰性銀屑病、銀屑病關節炎和尋常性銀屑病。2017年2月和2017年7月,Brodalumab分別又獲得美國和歐洲批準,用于對系統性療法或光照療法(紫外線治療)不響應的成人中重度斑塊狀銀屑病的治療。Brodalumab的安全有效性在3項安慰劑對照的三期臨床試驗中進行了研究,AMAGINE-1(NCT01708590),AMAGINE-2(NCT01708603)和AMAGINE-3(NCT01708603)三項試驗共納入患者4373名。主要終點是第12周銀屑病面積與嚴重程度改善達75%以上(PASI75)和醫生靜態總體評估(sPGA)得分(0/1)比基線至少下降2分的患者比例。AMAGINE-1結果顯示,Brodalumab 210mg 達PASI75的比例為83%,140mg組為60%,而安慰劑組為3%。sPGA得分達標率同樣高于安慰劑組,分別為76%,54%和1%。AMAGINE-2和AMAGINE-3試驗數據顯示,本品對中重度銀屑病臨床癥狀改善情況明顯優于安慰劑組。AMAGINE-2中210mg 組PASI達標率為86%,140mg組為67%,而安慰劑組為8%,AMAGINE-3則依次為86%,69%和6%,本品治療組sPGA達標率同樣更高,AMAGINE-2中210mg組達標率為79%,140mg組為58%,安慰劑組為4%; AMAGINE-3中,則依次為80%,60%和4%。
Avelumab
Avelumab (Bavencio,MSB0010718C)是一種以PD-L1為靶點的人IgG1單抗,首次批準于2017年5月23日。獲批適應癥為成人和12歲以上兒童的轉移性默克爾細胞癌。2017年9月,該適應癥也在歐洲獲批。Avelumab的獲批是基于二期關鍵性臨床試驗Javelin Merkel 200研究(NCT02155647) 的數據,該試驗包括兩部分,Part A患者事先經過化療,Part B患者則使用Avelumab初次治療。在10mg/kg的劑量下,每兩周一次靜注。Part A中,88名事先經過化療的患者使用本品治療后病情得到了控制,中位隨訪期為10.4個月,客觀緩解率為31.8%,其中8%完全緩解,20%部分緩解。Part B中,39名使用本品首次治療的患者,主要終點中位緩解時間的數據有望在2019年9月獲得。此前,Avelumab在歐洲和澳大利亞均獲得了默克爾細胞癌的孤兒藥地位,還獲得FDA突破性療法認定。2017年5月9日,FDA授予avelumab用于經鉑化療后進展的局部晚期或轉移性尿路上皮癌適應癥優先審評。除此以外,本品用于非小細胞肺癌、腎細胞癌、卵巢癌和胃癌治療的4項三期臨床試驗將在2018年達到終點,而且被美國和歐洲雙雙授予胃癌的孤兒藥資格。
Dupilumab
Dupilumab (Dupixent,REGN668/SAR231893)是一種以IL4R為靶點的IgG4單抗,于2017年4月28日和2017年9月28日分別獲得FDA和EMA批準用于特應性皮炎治療。Dupixent的獲批是基于一項名為LIBERTY的特應性皮炎研究項目,該項目包括SOLO1,SOLO2和CHRONOS三項臨床試驗。在SOLO1和SOLO2中,患者分別每周一次接受300mg Dupixent或安慰劑治療,或每兩周一次接受300mg Dupixent治療后,換用安慰劑治療。CHRONOS試驗則是以安慰劑為對照研究Dupixent與皮質類固醇聯合用藥的療效。試驗的主要終點為16周時研究者整體評價法(IGA)得分(0/1)比基線下降大于2分的患者比例。結果顯示,Dupixent單藥治療16周,IGA達標情況顯著高于安慰劑組,SOLO1為37%,SOLO2為36%。CHRONOS的試驗結果與此相似,dupilumab+皮質類固醇聯合治療組為39%,而安慰劑組+類固醇組為12%。因為療效卓著,本品獲得FDA的特應性皮炎的突破性療法認定。除特應性皮炎之外,dupilumab還在開展哮喘和鼻息肉方面的適應癥,目前處在三期臨床階段。2017年9月,賽諾菲/再生元宣布dupilumab用于無法控制的持續性哮喘的LIBERTY ASTHMA QUEST (NCT02414854)試驗達到治療終點。另外兩項關于鼻息肉治療的臨床試驗(NCT02912468, NCT02898454)也將在2018年達到終點。更可喜的是本品還能用于嗜酸性食道炎治療,目前研究處于臨床二期階段,而且還獲得了FDA的孤兒藥認定。
Ocrelizumab
Ocrelizumab (Ocrevus)是一種人源化的IgG1單抗,作用靶點是CD-20陽性的B細胞。這種B細胞可通過產生促炎性細胞因子、分泌自身抗體和活化促炎性T細胞,而在髓鞘損傷和多發性硬化癥發病中發揮作用。Ocrevus主要通過三種方式消滅CD20陽性B細胞:補體依賴的細胞毒作用和抗體依賴的細胞介導的細胞毒作用,以及在靶細胞上與CD20結合而使其凋亡。這種單抗在內風濕性關節炎(RA)和系統性紅斑狼瘡(SLE)上都進行了臨床研究,但預期效果不理想。Ocrevus于3月29日獲得FDA批準用于復發緩解型的多發性硬化以及原發進展型多發性硬化治療,此前該適應癥獲得了FDA突破性療法認定、快速通道和優先審評資格。OPERAI(NCT01247324)和OPERAII(NCT01412333)兩項試驗對Ocrevus治療發緩解型的多發性硬化的安全和有效性分別進行了評估,結果顯示Ocrevus治療組的年華復發率顯著低于β干擾素組,其中OPERAI低46%,而OPERAII低57%。原發進展型多發性硬化方面,ORATORIO試驗的結果顯示,與安慰劑相比,Ocrevus使12周(主要終點)的臨床殘疾進展顯著降低(32.9% vs 39.3%)。另外MRI顯示,接受Ocrevus治療的患者腦病變的總體積下降3.4%,而安慰劑增加7.4%。
Durvalumab
Durvalumab (Imfinzi,MEDI4736)是一種人IgG1抗體,可直接激活PD-L1以阻斷PD-L1及其受體(PD-1, CD80)間的聯系。Durvalumab設計初衷是阻斷細胞毒性效應與PD-L1陽性免疫細胞間的作用,于2017年5月1日獲FDA加速批準用于鉑化療惡化的局部晚期或轉移性尿路上皮癌。這是一個集優先審評和突破性療法于一身的產品,此次獲批是基于一項182名患者參與的1/2期臨床試驗(NCT01693562)。以10mg/kg的劑量每二周一次靜脈注射Durvalumab,連續用藥12個月,PD-L1高表達亞組的客觀緩解率為27.4%,PD-L1低表達亞組為4.1%。此外,鉑化療進展后局部晚期或無法手術治療的非小細胞肺癌(NSCLC)適應癥NDA已進入FDA審評階段,此前該申請獲得了FDA優先審評和突破性療法認定。與此同時,該適應癥的上市許可申請(MAA)也獲得EMA受理,三期PACIFI臨床試驗(NCT02125461)的數據也已一同提交到EMA。臨床結果顯示化療后進展的NSCLC,以化療為背景添加本品治療,中位無進展生存期為16.8月,而安慰劑只有5.6月。除以上兩個適應癥外,Durvalumab治療PD-L1陽性的轉移性頭頸癌還被FDA授予快速通道資格,三期臨床試驗EAGLE(NCT02369874)和KESTREL(NCT02551159)有望在2018年2月和3月分別達到終點。
Sarilumab
Sarilumab (Kevzara,SAR153191,REGN88)是一種以IL-6R為靶點的IgG1單抗,2017年2月在加拿大首次獲批,2017年5月和2017年7月分別又獲FDA和EMA批準用于對一種或多種生物制品或非生物制品疾病調節性抗風濕藥物(DMARDs)不充分響應或不耐受的中重度活動性類風濕性關節炎。Sarilumab的獲批是基于一項全球多中心的SARIL-RA臨床研究項目,其包括MOBILITY (NCT01061736),TARGET (NCT01709578)和MONARCH (NCT02332590) 三項三期臨床試驗。在MOBILITY和TARGET試驗中,sarilumab均與甲氨蝶呤或其他DMARD合用。MOBILITY研究結果顯示,200mg sarilumab治療組24周ACR20達66%,150mg組為58%,而安慰劑組僅33%。Sarilumab同樣可改善患者生理功能,200mg sarilumab治療組HAQ-DI評分下降0.58,150mg組為0.54,安慰劑組為0.30。TARGET試驗結果與前者相似,DMARD+200mg sarilumab治療組 ACR20達61%,150mg組為56%,而安慰劑組為34%。生理功能改善方面,DMARD+200mg sarilumab治療組HAQ-DI評分下降0.50,150mg組下降0.49,而安慰劑組僅0.29。MONARCH試驗則以阿達木單抗為對照評估sarilumab的安全有效性。24周治療結果顯示,使用紅細胞沉降率計算的28個關節的疾病活動度評分(DAS28-ESR)從基線下降3.28,而阿達木單抗組則只下降2.20,該試驗也達到主要終點。
Guselkumab
Guselkumab (Tremfya) 是一種人IgG1λ單抗,能夠阻滯IL-23的p19亞基,從而限制炎癥響應。IL-23是一種參與Th17細胞分化和維持的細胞因子,通過T細胞亞群生成促炎性細胞因子IL-17、IL-22和TNF。2017年7月13日,guselkumab獲得FDA批準用于適合系統療法(注射或口服治療)或光治療(紫外線治療)的中重度斑塊型銀屑病治療,2017年11月,guselkumab再次獲得EMA批準,適應癥與美國相同。Guselkumab獲批是基于三項臨床試驗的結果,分別為VOYAGE1(NCT02207231),VOYAGE2(NCT02207244)和NAVIGATE(NCT02203032)。其中VOYAGE1和VOYAGE2評估了Tremfya相對于安慰劑和阿達木單抗的療效和安全性,結果顯示,guselkumab治療16周,IGA達標率遠高于安慰劑組(85.1% vs 6.9%),另外,Tremfya治療組分別有73.3%和70.0%的患者實現PASI90 緩解,而阿達木單抗治療組分別為49.7%和46.8%。NAVIGATE試驗則評估了Tremfya相對于Stelara的療效和安全性,患者在0、4周接受ustekinumab治療,16周后響應不足的患者分別再次給予ustekinumab或guselkumab治療,主要終點是28周的IGA評分相比基線下降最低2分的患者比例。而結果顯示經ustekinumab或guselkumab治療,28周IGA評分達標率分別為14.3%和31.1%,32周時則分別為17.3%和36.3%,數據說明Tremfya在既往接受Stelara治療應答不足的患者中仍具有顯著的療效。
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa,CMC-544)是一種抗CD22的人源化IgG4抗體與細胞毒類藥物卡奇霉素的偶聯物。Besponsa于2017年6月30日在歐洲首次獲批,2017年8月17日再次獲得FDA批準,適應癥均為成人復發性或難治性B細胞前體急性淋巴細胞白血病(ALL)。在此此前,Besponsa已經獲得歐美雙方的孤兒藥資格,和FDA的ALL突破性療法認定。Besponsa的獲批是基于一項名為INOVATE ALL三期臨床試驗(NCT01564784),該試驗以現有的標準療法進行對照。患者在第1天(0.8mg/m2)、第8天(0.5mg/m2)和第15天(0.5mg/m2)分別接受三個劑量的Besponsa治療或標準化療。起始療程為21天,此后每個療程均為28天,直達6個療程。結果顯示Besponsa組患者完全緩解率為80.7%,中位總生存期為7.7個月,標準化療組完全緩解率為29.4%,中位總生存期為6.7個月。
Benralizumab
Benralizumab (Fasenra,MEDI-563) 是一個去巖藻糖基化的IgG1單抗,靶點為IL-5R的α亞基。 FDA于2017年11月14日批準本品用于12 歲以上12歲及以上具有嗜酸性表型的重度哮喘 患者的附加維持治療,2017年11月10日,EMA的專家委員會也對本品的上市給出積極的意見。本品的安全有效性WINDWARD研究項目中得到評估,該項目包括SIROCCO,CALIMA,ZONDA,BISE,BORA和GREGALE等6項三期臨床試驗。其中SIROCCO(NCT01928771)和CALIMA (NCT01914757)兩項隨機、雙盲、平行、安慰劑控制的臨床試驗評價了本品長期用藥安全性,患者長期以30mg的固定劑量皮下注射benralizumab長達56周,研究的結果在2016年公開。在為期28周的ZONDA(NCT02075255)試驗中,中重度哮喘患者在口服糖皮質激素的基礎上每四周或8周一次注射Benralizumab 30mg 或安慰劑對病情維持治療,結果顯示,Benralizumab組糖皮質激素用量降低75%,而安慰劑組僅下降 25%。
Emicizumab
Emicizumab (Hemlibra,emicizumab-kxwh,ACE910,RO5534262) 是一種雙特異性IgG4抗體,靶點為IXa和X,2017年11月16日首次獲得FDA批準用于預防或減少體內含有凝血因子VIII抑制物的A型血友病患者的出血頻率。在此之前,Emicizumab已經獲得FDA的孤兒藥和突破性療法認定。日本和歐洲的上市許可申請審評中,歐洲被授予加速審評資格,日本則給予孤兒藥認定。Emicizumab最早由中外發現,與羅氏一同開發并推向全球推向市場。本品的獲批是基于HAVEN1(NCT02622321)的臨床研究結果和HAVEN2(NCT02795767)的中期結果。HAVEN1研究表明,12歲及以上的體內含有VIII因子抑制物的A型血友病患者在接受Hemlibra預防治療后,與沒有接受預防患者相比,年出血率顯著降低87%(2.9 vs 23.3)。AVEN2研究的中期結果表明,12歲以下的體內含有抑制物的A型血友病兒童患者,在接受Hemlibra預防后,有87%未出現出血。在參加NIS 的13名兒童患者的患者內分析中,Hemlibra預防治療與接受旁路制劑治療(BPA)的患者相比出血率降低99%。
處于歐美審評中的單抗
截止2017年12月1日,一共有9個單抗產品在歐美的審評中,除 mogamulizumab于2012年已經在日本獲批外,ibalizumab,burosumab,tildrakizumab,caplacizumab,erenumab,fremanezumab,galcanezumab,romosozumab 尚未在任何國家批準。這些產品有望在2018年問世,其中burosumab和ibalizumab有望在1月或2月獲批上市。
2017年EMA/FDA審評中的單抗藥物

Ibalizumab
Ibalizumab是一種以CD4為靶點的IgG4單抗,目前處于FDA審評中,申請適應癥為多種抗病毒藥物治療耐藥的HIV感染。這是一個集孤兒藥資格和突破性療法于一身的藥物,因為手握FDA的優先審評資格,PDUFA期限至2018年1月3日。用于支持BLA的是一項名為TMB-301(NCT02475629)的三期臨床試驗,該試驗開放標簽地研究了Ibalizumab的安全有效性。2017年10月,Thera technologies Inc宣布TMB301研究已經完成,并將繼續拓展研究項目TMB-311(NCT02707861)。27名完成TMB-301 24周試驗的患者被納入TMB-311研究,他們將每二周一次接受ibalizumab 800mg持續48周。在TMB-311試驗中,15名在24周未檢出病毒載量的患者,該狀態被保持至48周。其余在24周時能檢測到病毒載量的患者,17名(63%)在48周時病毒載量下降至200拷貝/ml以下。
Burosumab
Burosumab (KRN23)是一種人IgG1單抗,靶點為成纖維細胞生長因子23(FGF23)。Burosumab由日本麒麟公司發現,申請適應癥為家族性低磷酸鹽血癥佝僂病(XLH)。FGF23是一種激素,它與腎臟磷分泌控制和活性維生素D生成相關。這種病的特點主要是FGF23水平過剩造成骨骼肌缺陷,以及腫瘤導致的骨軟化。Burosumab的上市申請已經同時提交到FDA和EMA,在美國已經獲得XLH突破性療法認定和優先審評資格,PDUFA至2018年4月17日,歐洲方面,本品已經在2017年12月14日獲得CHMP的積極意見。在一項隨機、雙盲、安慰劑對照的三期臨床試驗(NCT02526160)中,Burosumab可顯著增加XLH患者的血磷水平,治療達到主要終點。試驗中,35名XLH患者4周一次隨機接受1mg/kg的burosumab或安慰劑治療24周,結果顯示94%的burosumab治療組患者(n=64)血磷水平超過正常下限,而且持續24周維持血磷水平在正常范圍內,而相比之下,安慰劑血磷達正常范圍的患者僅8%。另一項burosumab與口服磷/活性維生素D聯合治療兒童XLH的臨床試驗(NCT02915705)將在2018年7月完成。
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222)是一種人源化的IgG1單抗,靶點為IL-23p19,用于中重度斑塊銀屑病的上市申請已經提交到FDA和EMA。支持BLA的數據來于兩項三期臨床試驗(reSURFACE1/2),在這兩項試驗中,共1800余名患者被納入研究,部分患者治療周期長達3.5年。為期52周、安慰劑對照、平行設計的reSURFACE2試驗(NCT01729754)比較了Tildrakizumab(100/200mg)相對依那西普的安全性和耐受性。reSURFACE1試驗(NCT017223310)與reSURFACE2的設計相似,但加入了活性藥物對照組。在reSURFACE2試驗的12周,tildrakizumab 200mg組和100mg組達PASI75的患者比例分別為66%和61%,顯著高于依那西普治療組的48%和安慰劑組的6%。tildrakizumab 200mg組和100mg的PGA達標率均為59%,而依那西普治療組為48%,安慰劑組為4%。reSURFACE1試驗也得到基本相似的結果。
Caplacizumab
Caplacizumab (ALX-0081) 是一個納米抗體,靶點為血管性血友病因子,申請適應癥為獲得性血栓性血小板減少性紫癜(aTTP)。這是一種罕見而嚴重的彌散性血栓性微血管病,以微血管病性溶血性貧血、血小板聚集消耗性減少,以及微血栓形成造成器官損害(如腎臟、中樞神經系統等)為特征。此前Caplacizumab已分別獲得FDA和EMA的aTTP孤兒藥認定,FDA還給予了快速審評通道支持。2017年2月,Ablynx宣布MAA已經提交到EMA,aTTP的三期臨床試驗HERCULES數據已經獲得,產品的安全性和有效性得到了確認。該試驗共招募了145名急性aTTP患者,患者被1:1隨機分配到caplacizumab組或安慰劑組,所有患者給予標準護理,即每日血漿置換和免疫抑制劑。患者在血漿置換后的第一天被靜注10mg的caplacizumab或安慰劑,然后每日皮下注射caplacizumab或安慰劑,連續治療30天。根據患者的應答情況,在30天的基礎上再加一個7-28天的額外治療期,主要終點為血小板計數響應所需時間。研究結果顯示,caplacizumab治療組患者的血小板計數響應時間比安慰劑發生統計學意義上的顯著減少,患者達到血小板計數響應的概率增加50%,試驗期間患者死亡率、aTTP復發率、一次以上血栓事件的發生率綜合降低74%,整個研究期間的aTTP復發率降低67%。完成HERCULES試驗后的患者已經被納入為期三年的follow-up試驗(NCT02878603)中,該試驗目前尚在進行中。
Erenumab
Erenumab (Aimovig,AMG334)是一種人IgG2單抗,靶點為降鈣素基因相關肽(CGRP),而CGRP被認為與感受神經元敏化和受損情況進展相關,申請適應癥為偏頭痛,目前上市申請已經提交到FDA和EMA。本品由安進和諾華共同開發,美國的經營權歸安進公司所有,PDUFA期限至2018年5月17日,歐洲和除日本以外的其它地區,經營權歸諾華所有。支持BLA的數據為4項二期或三期臨床試驗的研究結果。超過2600名每月頭痛超過4次的患者加入了臨床試驗,這些試驗證明erenumab相比安慰劑可顯著減少患者每月頭痛的天數,顯著降低頭痛致殘或急性發作用藥治療的次數。一項關于陣發性偏頭痛的臨床試驗NCT02456740研究結果顯示,955名參與研究的患者每月平均偏頭痛發作天數為8.3天,試驗4-6個月后,70mg Erenumab組月均偏頭痛減少3.2天,140mg Erenumab組月均偏頭痛減少3.7天,而安慰劑組月均偏頭痛減少1.8天。70mg Erenumab組43.4%的患者以及140mg Erenumab組50.0%的患者偏頭痛發作天數減少50%及以上,而安慰劑組該終點達標率為26.6%;70mg Erenumab組機體功能損傷得分減少4.2分,140mg Erenumab組減少4.8分,而安慰劑組減少2.4分;70mg Erenumab組日常活動得分改善5.5分,140mg Erenumab組改善5.9分,而安慰劑組改善3.3分。
Fremanezumab
Fremanezumab (TEV-48125) 是一種IgG2單抗,靶點為降鈣素基因相關肽(CGRP),適應癥也是偏頭痛。支持本品BLA的數據包括HALO研究項目在內的三期臨床試驗數據,臨床試驗納入了2000多名陣發性偏頭痛或慢性偏頭痛患者,而且試驗的主要終點和次要終點都已經達到。納入HALO研究項目的陣發性偏頭痛患者按1:1:1分隨機三組,一組皮下給予fremanezumab 225mg/月,持續3個月,另一組起始給予fremanezumab 675mg,隨后的2個月給予安慰劑,第三組則給予相應的安慰劑。試驗的治療終點為12周時,患者每月頭痛天數相對基線的改變情況。結果顯示按月給藥方案,患者每月偏頭痛天數相對基線顯著下降41.6%(-3.7天 vs -2.2天),按季度給藥方案,每月偏頭痛次數降低3.4天或37.0%。納入HALO研究項目的慢性偏頭痛患者同樣按1:1:1分隨機三組,一組皮下給予fremanezumab 225mg/月,持續3個月,另一組起始給與fremanezumab 675mg,隨后的2個月給予安慰劑,第三組則給予相應的安慰劑。結果顯示,患者12周內每月偏頭痛天數相對安慰劑下降2.5天,按月給藥方案每月頭痛天數降低4.6天,按季度給藥方案每月頭痛天數下降4.3天。
Galcanezumab
Galcanezumab (LY2951742) 是一種IgG4單抗,靶點為降鈣素基因相關肽(CGRP),適應癥還是偏頭痛。 支持BLA的臨床數據來自3項三期臨床試驗EVOLVE-1,EVOLVE-2和REGAIN。在EVOLVE-1中,120mg galcanezumab組患者每月頭痛天數相對基線下降50%的比例為62.3%,240mg組為60.9%,安慰劑組為38.6%,120mg galcanezumab組患者每月頭痛天數相對基線下降75%的比例為38.8%,240mg組為38.5%,安慰劑組為19.3%;120mg galcanezumab組患者每月頭痛天數相對基線下降100%的比例為15.6%,240mg組為14.6%,安慰劑組為6.2%。EVOLVE-2試驗也得到相似的結果數據。在安慰劑對照的REGAIN試驗中,慢性偏頭痛患者被給予120mg或240mg的galcanezumab,持續3個月以上治療,患者每月平均頭痛天數顯著低于對照組。除此以外,本品還在進行安慰劑對照的慢性叢集性頭痛三期臨床研究(NCT02438826)。患者每月一次皮下注射galcanezumab,連續治療12個月,主要結果指標是每周叢集性頭痛發作頻率相對基線的改善,該臨床試驗已經招募了162名受試者,試驗結果有望在2018年3月揭曉。此前,該適應癥已獲得FDA的快速審評通道資格,該認證有望加速叢集性頭痛適應癥的上市速度。
Romosozumab
Romosozumab (EVENITYTM,AMG785) 是一種人源化IgG2單抗,靶點為硬骨素,romosozumab的BLA已經于2016年7月提交到FDA,申請適應癥為骨質疏松。支持BLA的數據包括安慰劑對照的FRAME(NCT01575834)三期臨床研究結果。受試者每月一次接受romosozumab 210mg皮下注射,持續至第12個月,然后每半年一次皮下注射romosozumab 60mg,持續至第24個月。隨后三期臨床試驗 ARCH(NCT01631214)結果顯示,每月一次皮下注射本品210mg與每周一次口服阿侖膦酸鈉相比,嚴重心血管不良事件大幅增加(2.5% vs 1.9%),2017年7月,安進公司宣布收到FDA的完全回信,要求其提供ARCH的安全性數據和三期臨床試驗BRIDGE(NCT02186171)數據,以評估男性骨質疏松患者的用藥安全有效性。
Mogamulizumab
Mogamulizumab (KW-0761,Poteligeo)是一種去巖藻糖基的人源化IgG1單抗,靶點為表達在皮膚T細胞淋巴瘤患者腫瘤細胞上的CC趨化因子受體4(CCR4)。本品已經在2012年3月獲得日本PMDA批準用于復發或難治性CCR4陽性的成人T細胞白血病(ATL)治療,2014年3月獲準用于CCR4陽性的周圍T細胞淋巴瘤和皮膚T細胞淋巴瘤治療,2014年12月又獲準用于未經化療的CCR4陽性成人T細胞白血病治療。2017年7月,日本麒麟公司宣布mogamulizumab用于至少經過一種藥物系統性治療后復發的T細胞淋巴瘤治療的上市申請已經處于EMA的審評中。支持MAA的數據是一項隨機、開放標簽、多中心的三期臨床試驗MAVORIC (NCT01728805),試驗以vorinostat為對照評估了mogamulizumab的安全有效性。372名難治性CTCL患者隨機接受vorinostat或mogamulizumab,2017年4月麒麟公司宣布,MAVORIC試驗達到既定的PFS臨床終點(254天vs169天),2017年8月,mogamulizumab獲得FDA的蕈樣肉芽腫和塞扎里綜合征突破性療法認定。
將在2018提交上市申請的單抗
已經獲得或近期即將獲得關鍵性臨床終點數據,在2018年有望提交BLA的單抗藥物有12個,分別為lanadelumab, crizanlizumab, ravulizumab, eptinezumab, risankizumab, satralizumab, brolucizumab, PRO140, sacituzumab govitecan, moxetumomab pasudotox, cemiplimab和ublituximab。根據FDA的審評周期,這12個藥物也有望在2018年底或2019年上市,其中前8個為一般藥物,后4個是抗癌藥。
Ibalizumab是一種以CD4為靶點的IgG4單抗,目前處于FDA審評中,申請適應癥為多種抗病毒藥物治療耐藥的HIV感染。這是一個集孤兒藥資格和突破性療法于一身的藥物,因為手握FDA的優先審評資格,PDUFA期限至2018年1月3日。用于支持BLA的是一項名為TMB-301(NCT02475629)的三期臨床試驗,該試驗開放標簽地研究了Ibalizumab的安全有效性。2017年10月,Thera technologies Inc宣布TMB301研究已經完成,并將繼續拓展研究項目TMB-311(NCT02707861)。27名完成TMB-301 24周試驗的患者被納入TMB-311研究,他們將每二周一次接受ibalizumab 800mg持續48周。在TMB-311試驗中,15名在24周未檢出病毒載量的患者,該狀態被保持至48周。其余在24周時能檢測到病毒載量的患者,17名(63%)在48周時病毒載量下降至200拷貝/ml以下。
Burosumab
Burosumab (KRN23)是一種人IgG1單抗,靶點為成纖維細胞生長因子23(FGF23)。Burosumab由日本麒麟公司發現,申請適應癥為家族性低磷酸鹽血癥佝僂病(XLH)。FGF23是一種激素,它與腎臟磷分泌控制和活性維生素D生成相關。這種病的特點主要是FGF23水平過剩造成骨骼肌缺陷,以及腫瘤導致的骨軟化。Burosumab的上市申請已經同時提交到FDA和EMA,在美國已經獲得XLH突破性療法認定和優先審評資格,PDUFA至2018年4月17日,歐洲方面,本品已經在2017年12月14日獲得CHMP的積極意見。在一項隨機、雙盲、安慰劑對照的三期臨床試驗(NCT02526160)中,Burosumab可顯著增加XLH患者的血磷水平,治療達到主要終點。試驗中,35名XLH患者4周一次隨機接受1mg/kg的burosumab或安慰劑治療24周,結果顯示94%的burosumab治療組患者(n=64)血磷水平超過正常下限,而且持續24周維持血磷水平在正常范圍內,而相比之下,安慰劑血磷達正常范圍的患者僅8%。另一項burosumab與口服磷/活性維生素D聯合治療兒童XLH的臨床試驗(NCT02915705)將在2018年7月完成。
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222)是一種人源化的IgG1單抗,靶點為IL-23p19,用于中重度斑塊銀屑病的上市申請已經提交到FDA和EMA。支持BLA的數據來于兩項三期臨床試驗(reSURFACE1/2),在這兩項試驗中,共1800余名患者被納入研究,部分患者治療周期長達3.5年。為期52周、安慰劑對照、平行設計的reSURFACE2試驗(NCT01729754)比較了Tildrakizumab(100/200mg)相對依那西普的安全性和耐受性。reSURFACE1試驗(NCT017223310)與reSURFACE2的設計相似,但加入了活性藥物對照組。在reSURFACE2試驗的12周,tildrakizumab 200mg組和100mg組達PASI75的患者比例分別為66%和61%,顯著高于依那西普治療組的48%和安慰劑組的6%。tildrakizumab 200mg組和100mg的PGA達標率均為59%,而依那西普治療組為48%,安慰劑組為4%。reSURFACE1試驗也得到基本相似的結果。
Caplacizumab
Caplacizumab (ALX-0081) 是一個納米抗體,靶點為血管性血友病因子,申請適應癥為獲得性血栓性血小板減少性紫癜(aTTP)。這是一種罕見而嚴重的彌散性血栓性微血管病,以微血管病性溶血性貧血、血小板聚集消耗性減少,以及微血栓形成造成器官損害(如腎臟、中樞神經系統等)為特征。此前Caplacizumab已分別獲得FDA和EMA的aTTP孤兒藥認定,FDA還給予了快速審評通道支持。2017年2月,Ablynx宣布MAA已經提交到EMA,aTTP的三期臨床試驗HERCULES數據已經獲得,產品的安全性和有效性得到了確認。該試驗共招募了145名急性aTTP患者,患者被1:1隨機分配到caplacizumab組或安慰劑組,所有患者給予標準護理,即每日血漿置換和免疫抑制劑。患者在血漿置換后的第一天被靜注10mg的caplacizumab或安慰劑,然后每日皮下注射caplacizumab或安慰劑,連續治療30天。根據患者的應答情況,在30天的基礎上再加一個7-28天的額外治療期,主要終點為血小板計數響應所需時間。研究結果顯示,caplacizumab治療組患者的血小板計數響應時間比安慰劑發生統計學意義上的顯著減少,患者達到血小板計數響應的概率增加50%,試驗期間患者死亡率、aTTP復發率、一次以上血栓事件的發生率綜合降低74%,整個研究期間的aTTP復發率降低67%。完成HERCULES試驗后的患者已經被納入為期三年的follow-up試驗(NCT02878603)中,該試驗目前尚在進行中。
Erenumab
Erenumab (Aimovig,AMG334)是一種人IgG2單抗,靶點為降鈣素基因相關肽(CGRP),而CGRP被認為與感受神經元敏化和受損情況進展相關,申請適應癥為偏頭痛,目前上市申請已經提交到FDA和EMA。本品由安進和諾華共同開發,美國的經營權歸安進公司所有,PDUFA期限至2018年5月17日,歐洲和除日本以外的其它地區,經營權歸諾華所有。支持BLA的數據為4項二期或三期臨床試驗的研究結果。超過2600名每月頭痛超過4次的患者加入了臨床試驗,這些試驗證明erenumab相比安慰劑可顯著減少患者每月頭痛的天數,顯著降低頭痛致殘或急性發作用藥治療的次數。一項關于陣發性偏頭痛的臨床試驗NCT02456740研究結果顯示,955名參與研究的患者每月平均偏頭痛發作天數為8.3天,試驗4-6個月后,70mg Erenumab組月均偏頭痛減少3.2天,140mg Erenumab組月均偏頭痛減少3.7天,而安慰劑組月均偏頭痛減少1.8天。70mg Erenumab組43.4%的患者以及140mg Erenumab組50.0%的患者偏頭痛發作天數減少50%及以上,而安慰劑組該終點達標率為26.6%;70mg Erenumab組機體功能損傷得分減少4.2分,140mg Erenumab組減少4.8分,而安慰劑組減少2.4分;70mg Erenumab組日常活動得分改善5.5分,140mg Erenumab組改善5.9分,而安慰劑組改善3.3分。
Fremanezumab
Fremanezumab (TEV-48125) 是一種IgG2單抗,靶點為降鈣素基因相關肽(CGRP),適應癥也是偏頭痛。支持本品BLA的數據包括HALO研究項目在內的三期臨床試驗數據,臨床試驗納入了2000多名陣發性偏頭痛或慢性偏頭痛患者,而且試驗的主要終點和次要終點都已經達到。納入HALO研究項目的陣發性偏頭痛患者按1:1:1分隨機三組,一組皮下給予fremanezumab 225mg/月,持續3個月,另一組起始給予fremanezumab 675mg,隨后的2個月給予安慰劑,第三組則給予相應的安慰劑。試驗的治療終點為12周時,患者每月頭痛天數相對基線的改變情況。結果顯示按月給藥方案,患者每月偏頭痛天數相對基線顯著下降41.6%(-3.7天 vs -2.2天),按季度給藥方案,每月偏頭痛次數降低3.4天或37.0%。納入HALO研究項目的慢性偏頭痛患者同樣按1:1:1分隨機三組,一組皮下給予fremanezumab 225mg/月,持續3個月,另一組起始給與fremanezumab 675mg,隨后的2個月給予安慰劑,第三組則給予相應的安慰劑。結果顯示,患者12周內每月偏頭痛天數相對安慰劑下降2.5天,按月給藥方案每月頭痛天數降低4.6天,按季度給藥方案每月頭痛天數下降4.3天。
Galcanezumab
Galcanezumab (LY2951742) 是一種IgG4單抗,靶點為降鈣素基因相關肽(CGRP),適應癥還是偏頭痛。 支持BLA的臨床數據來自3項三期臨床試驗EVOLVE-1,EVOLVE-2和REGAIN。在EVOLVE-1中,120mg galcanezumab組患者每月頭痛天數相對基線下降50%的比例為62.3%,240mg組為60.9%,安慰劑組為38.6%,120mg galcanezumab組患者每月頭痛天數相對基線下降75%的比例為38.8%,240mg組為38.5%,安慰劑組為19.3%;120mg galcanezumab組患者每月頭痛天數相對基線下降100%的比例為15.6%,240mg組為14.6%,安慰劑組為6.2%。EVOLVE-2試驗也得到相似的結果數據。在安慰劑對照的REGAIN試驗中,慢性偏頭痛患者被給予120mg或240mg的galcanezumab,持續3個月以上治療,患者每月平均頭痛天數顯著低于對照組。除此以外,本品還在進行安慰劑對照的慢性叢集性頭痛三期臨床研究(NCT02438826)。患者每月一次皮下注射galcanezumab,連續治療12個月,主要結果指標是每周叢集性頭痛發作頻率相對基線的改善,該臨床試驗已經招募了162名受試者,試驗結果有望在2018年3月揭曉。此前,該適應癥已獲得FDA的快速審評通道資格,該認證有望加速叢集性頭痛適應癥的上市速度。
Romosozumab
Romosozumab (EVENITYTM,AMG785) 是一種人源化IgG2單抗,靶點為硬骨素,romosozumab的BLA已經于2016年7月提交到FDA,申請適應癥為骨質疏松。支持BLA的數據包括安慰劑對照的FRAME(NCT01575834)三期臨床研究結果。受試者每月一次接受romosozumab 210mg皮下注射,持續至第12個月,然后每半年一次皮下注射romosozumab 60mg,持續至第24個月。隨后三期臨床試驗 ARCH(NCT01631214)結果顯示,每月一次皮下注射本品210mg與每周一次口服阿侖膦酸鈉相比,嚴重心血管不良事件大幅增加(2.5% vs 1.9%),2017年7月,安進公司宣布收到FDA的完全回信,要求其提供ARCH的安全性數據和三期臨床試驗BRIDGE(NCT02186171)數據,以評估男性骨質疏松患者的用藥安全有效性。
Mogamulizumab
Mogamulizumab (KW-0761,Poteligeo)是一種去巖藻糖基的人源化IgG1單抗,靶點為表達在皮膚T細胞淋巴瘤患者腫瘤細胞上的CC趨化因子受體4(CCR4)。本品已經在2012年3月獲得日本PMDA批準用于復發或難治性CCR4陽性的成人T細胞白血病(ATL)治療,2014年3月獲準用于CCR4陽性的周圍T細胞淋巴瘤和皮膚T細胞淋巴瘤治療,2014年12月又獲準用于未經化療的CCR4陽性成人T細胞白血病治療。2017年7月,日本麒麟公司宣布mogamulizumab用于至少經過一種藥物系統性治療后復發的T細胞淋巴瘤治療的上市申請已經處于EMA的審評中。支持MAA的數據是一項隨機、開放標簽、多中心的三期臨床試驗MAVORIC (NCT01728805),試驗以vorinostat為對照評估了mogamulizumab的安全有效性。372名難治性CTCL患者隨機接受vorinostat或mogamulizumab,2017年4月麒麟公司宣布,MAVORIC試驗達到既定的PFS臨床終點(254天vs169天),2017年8月,mogamulizumab獲得FDA的蕈樣肉芽腫和塞扎里綜合征突破性療法認定。
將在2018提交上市申請的單抗
已經獲得或近期即將獲得關鍵性臨床終點數據,在2018年有望提交BLA的單抗藥物有12個,分別為lanadelumab, crizanlizumab, ravulizumab, eptinezumab, risankizumab, satralizumab, brolucizumab, PRO140, sacituzumab govitecan, moxetumomab pasudotox, cemiplimab和ublituximab。根據FDA的審評周期,這12個藥物也有望在2018年底或2019年上市,其中前8個為一般藥物,后4個是抗癌藥。
2018年有望提交上市申請的單抗
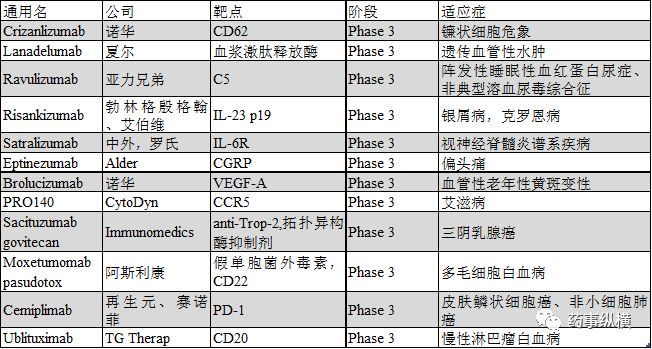
Lanadelumab
Lanadelumab (SHP643,DX-2930)是一種人IgG1單抗,可通過抑制血漿激肽釋放酶而阻止緩激肽的生產。Lanadelumab在三期臨床開展的適應癥為遺傳性血管性水腫(HAE),遺傳血管性水腫是一種以身體受到外界侵害后引發組織水腫為特征的罕見病。Lanadelumab已經獲得美國和歐洲授予的HAE孤兒藥資格和FDA HAE突破性療法認定。2017年5月,夏爾報告了一項為期26周的三期臨床HELP試驗(NCT02586805)數據,該試驗就lanadelumab的安全性和阻止 I 型和II型HAE患者急性血管水腫發生率的療效進行了評估。試驗中,49名患者每二周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 150mg或安慰劑。結果顯示,每二周一次給予lanadelumab 300mg,每四周一次給予lanadelumab 300mg和每四周一次給予lanadelumab 150mg的患者,血管水腫發生率相比安慰劑分別下降87%,73%和76%,試驗達到既定的主要終點和次要終點。另一項開放標簽的三期臨床長期安全有效性研究 (NCT02741596)用以評估lanadelumab阻止 I 型和II型HAE患者急性血管水腫發生的效率,治療終點將在2018年2月到達。本品的上市申請將在2018年初提交到FDA和EMA。
Crizanlizumab
Crizanlizumab (SEG101) 是一種人源化單抗,靶點為血小板選擇蛋白,也就是眾所周知的CD62,研究的適應癥為鐮狀細胞疼痛危象 (SCPC)。鐮狀細胞危象臨床表現為慢性溶血性貧血,易感染和再發性疼痛危象以致慢性局部缺血導致器官組織局部損壞。在安慰劑對照的二期SUSTAIN試驗(NCT01895361)中,198名16到65歲的患者,以5mg/kg的劑量,每四周一次給予crizanlizumab,連續治療50周,SCPC年化率中位值相對安慰劑下降45.3%(1.63 vs 2.98),用藥后首次發生SCPC的時間相比安慰劑延長2.9倍(中位時間分別為4.07月和1.38月),第二次發生SCPC的時間相比安慰劑延長2.0倍(中位時間分別為10.32月和5.09月)。此前,crizanlizumab在美國和歐洲均已獲得SCPC孤兒藥認定,如果PK/PD數據與最終的生產工藝生產的產品一致,諾華將在2018年提交BLA。
Ravulizumab
Ravulizumab (ALXN1210) 是一種人源化,以補體5(C5)為靶點的單抗,是eculizumab的下一代,目前處于注冊前期階段。其開展的臨床試驗包括2項與陣發性睡眠性血紅蛋白尿癥(PNH)相關的三期臨床試驗和2項與非典型溶血尿毒綜合征 (aHUS)相關的三期臨床試驗。這2項(PNH)三期臨床試驗NCT02946463 和NCT03056040是基于一項1/2期臨床試驗的結果。未經補體抑制劑治療的患者使用Ravulizumab后血漿乳酸脫氫酶(LDH)水平快速并持續下降,FACIT疲勞量表得分顯著改善。3期臨床試驗NCT02946463旨在評估Ravulizumab相對eculizumab治療未經補體抑制劑治療的成年PNH患者的安全有效性。該試驗中,患者基于體重在第一天給予一個負荷劑量,第15天給予維持劑量( 40至60kg體重:負荷劑量2400mg, 維持劑量每8周3000mg;60至100kg體重: 負荷劑量2700mg, 維持劑量每8周3300mg;體重大于100kg的患者,負荷劑量3000mg, 維持劑量每8周3600mg),而eculizumab則根據說明給藥。預期的臨床終點是26周內血漿乳酸脫氫酶(LDH)達正常水平的情況。試驗招募了246名患者,將在2017年12月達到終點。另一項3期臨床試驗則旨在評估Ravulizumab對使用eculizumab治療6個月以上且病情穩定患者的療效。Ravulizumab組的給藥方案與前一項臨床試驗相同,eculizumab組的患者則在第183天上給予負荷劑量的Ravulizumab,第197天給予維持劑量,主要終點是26周血漿乳酸脫氫酶(LDH)水平的改變情況。試驗招募了197名患者,試驗將在2018年3月結束。亞力兄弟有望在2018年2季度報告這兩項試驗的結果,并提交BLA。此前Ravulizumab已經獲得美國和歐洲PNH的孤兒藥認定。與aHUS相關的單臂三期臨床試驗NCT02949128招募了55名患者,試驗有望在2018年初結束,另一項關于兒童aHUS的三期臨床試驗NCT03131219也在進行中,結束時間是2018年12月。
Eptinezumab
Eptinezumab (ALD403) 是一個IgG1單抗,靶點為降鈣素基因相關肽(CGRP),開發適應癥是偏頭痛。2017年6月,Alder BioPharma宣布三期臨床 PROMISE1 試驗(NCT02559895)達到主要和次要終點。納入這項研究的陣發性偏頭痛患者平均頭痛天數是8.6天/月,試驗中患者分別給予eptinezumab 300mg、100mg、30mg或安慰劑,最終30mg的低劑量組未納入結果統計。結果顯示,1至12周觀察到患者每月頭痛次數顯著下降,300mg組每月頭痛為4.3天,100mg組為3.9天,安慰劑組為3.2天。接近三分之一的患者在第4-12周的每月頭痛天數下降75%以上,平均有五分之一的患者在1-6月內未至少有一個月未發生頭痛。另一項專門針對慢性偏頭痛設計的三期臨床試驗PROMISE2 (NCT02974153) 已招募1050名患者,該試驗將在2018年上半年結束,Alder計劃在2018年2季度提交上市申請。
Risankizumab
Risankizumab (ABBV066, BI655066)是一種以IL-23的p19亞基為靶點的IgG1單抗,開發適應癥為銀屑病。2017年10月,艾伯維宣布以ustekinumab和adalimumab為對照的三期臨床試驗達終點。在ultIMMa-1(NCT02684370)和ultIMMa-2(NCT02684357)試驗中,以ustekinumab和安慰劑為對照評估了risankizumab的有效性。經過16周的治療,兩項試驗中接受risankizumab治療的患者,達PASI90的患者比率均為75%,而接受ustekinumab患者,分別為 42%和48%,安慰劑則分別只有5%和2%。risankizumab治療組sPGA評分達標率分別為88%和84%,ustekinumab治療組分別為63%和62%,安慰劑組僅為8%和5%。在IMMvent研究(NCT02694523)中,患者分別接受risankizumab或adalimumab治療16周,risankizumab組達PASI90的患者比例為72%,阿達木單抗組則只有47%,risankizumab組sPGA達標率為84%,而阿達木單抗只有60%。本品有望在2018年提交BLA,2019年上市。
Satralizumab
Satralizumab (SA237) 是一種人源化IgG2單抗,靶點為白介素-6受體(IL-6R),開發適應癥為視神經脊髓炎或視神經脊髓炎譜系疾病。評估Satralizumab添加基線治療安全有效性的三期臨床試驗NCT02028884還在進行中,主要的治療終點是30個月內的疾病首次復發時間,該試驗已招募了70名患者,預期將于2018年7月結束。另一項安慰劑對照的三期臨床試驗NCT02073279也已經完成招募,主要的治療終點為38個月內疾病首次復發的時間,該試驗已經招募到98名患者,有望在2018年10月結束。此前該產品已經獲得FDA和EMA關于視神經脊髓炎譜系疾病的孤兒藥認定,有望在2018年下半年提交上市申請。
Brolucizumab
Brolucizumab (RTH258) 是一種單鏈可變區片段,靶點為血管表皮細胞生長因子A(VEGF)-A,開發適應癥為新生血管性老年性黃斑變性(nAMD)。2017年6月,諾華宣布三期臨床試驗HAWK (NCT02307682)和HARRIER (NCT02434328)達治療終點,Brolucizumab治療組48周平均最佳矯正視力相對基線改善情況非劣于aflibercept。這兩項試驗納入nAMD患者超過1800人,分別給予brolucizumab 6mg、brolucizumab 3mg(HARRIER 不涉及)或aflibercept 2mg。結果顯示Brolucizumab 6mg在兩項試驗中都以顯著p值到達主要終點和關鍵次要終點;Brolucizumab 3mg也在HAWK中到達這些終點。此外,在HAWK和HARRIER試驗中,12周用藥一次的患者分別有57%和52%堅持用藥到48周。Brolucizumab表現出良好的耐受性,總體眼睛和非眼睛不良反應發生率都與aflibercept相當。諾華有望在2018年完成低劑量規格的PK試驗,BLA有望在2018年下半年提交。
PRO140
PRO140 是一種人源化IgG4單抗,靶點為CC趨化因子受體5(CCR5),可防止HIV病毒進入T細胞,目前開發適應癥為艾滋病。兩項2/3期臨床試驗(NCT02483078和NCT02859961)已經在2017年10月和2017年12月完成。NCT02483078是一項隨機、雙盲、安慰劑對照的臨床試驗,耐藥的HIV感染者在最佳抗病毒藥的背景治療下,添加PRO140或安慰劑治療。而NCT02859961則研究的是抗逆轉錄病毒治療后病情處于穩定期的患者,換用PRO140單獨治療的有效性。2017年10月,CytoDyn與FDA進行會晤,就提交聯合用藥BLA所需的受試人數和患者類型展開商討,FDA要求NCT02483078試驗完成50例患者的研究,BLA要中提交總數300人的安全性評估。目前該產品已經獲得FDA快速審評通道資格,這也就意味著本品可以提交滾動BLA申請。
Sacituzumab govitecan
Sacituzumab govitecan (IMMU-132) 是一種抗體偶聯物,由人源化anti-Trop-2抗體偶聯伊立替康活性代謝物SN38而成,開發適應癥為晚期三陰乳腺癌 (TNBC)。2017年11月,Immunomedics宣布該公司將在2018年一季度提交BLA,此前本品已經獲得FDA三陰乳腺癌突破性療法認定,支持將BLA的數據來自NCT01631552研究。110例三陰乳腺癌患者接受10mg/kg的sacituzumab govitecan治療,客觀緩解率為34%,臨床受益率為46%,Kaplan-Meier(KM)中位有效期和無進展生存期分別為7.6月和5.5月。驗證本品對復發或難治性三陰乳腺癌療效的三期臨床試驗ASCENT(NCT02574455)已經招募到328名患者,入組的患者均為2種以上方案化療后進展或最少一種方案化療并接受輔助治療后12個月內進展的患者。試驗中患者分別接受sacituzumab govitecan或4個預先設定的單藥治療方案化療,該試驗已經在進行中。
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22,CAT- 8015) 是一種分子量為38kDa的重組銅綠假單胞菌外毒素的毒性部分與CD22抗體可變區片段融合的抗體偶聯物。早在2016年2月,Moxetumomab pasudotox就獲得FDA多毛細胞白血病 (HCL)的孤兒藥認定。本品目前在開展一項復發或難治性HCL的多中心、單臂三期臨床試驗(NCT01829711),該試驗已經在2017年五月達到主要終點,阿斯利康計劃在2018年提交BLA。
Cemiplimab
Cemiplimab (REGN2810, SAR439684) 是一種人程序性死亡受體1(PD-1)單抗,開發適應癥為無法手術的或轉移性皮膚鱗狀細胞癌(CSCC),目前該適應癥已經獲得FDA突破性療法認定。一期臨床試驗NCT02383212顯示本品對局部進展性CSCC和轉移性CSCC均有很好的療效,綜合客觀緩解率為46.2%。目前賽諾菲和再生元正在共同開發Cemiplimab,BLA有望在2018年一季度首次提交。另外一項以無法手術的或轉移性CSCC為研究對象,名為EMPOWER-CSCC1的二期臨床試驗也在開展中。除了CSCC,賽諾菲和再生元還在積極開展宮頸癌(NCT03257267)和非小細胞肺癌肺癌(NCT03088540)的三期臨床試驗,試驗終點將分別在2020年5月和2021年11月達到。
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101)是一種糖基化的嵌合型抗體,靶點為CD20。該抗體含低含量的巖藻糖,可改善FcγRIIIa的結合率,抗體依賴的細胞介導的細胞毒性作用(ADCC)相比利妥昔單抗有所增強,開發適應癥為慢性淋巴瘤白血病 (CLL)。一項多中心、開放標簽的三期臨床GENUINE試驗(NCT02301156)評估了ublituximab與依魯替尼聯合用藥相對依魯替尼單獨用藥的安全有效性。該試驗共有126名CLL患者入組,患者每日一次給予依魯替尼420mg或依魯替尼420mg+ublituximab,結果顯示聯合用藥組客觀緩解率為78%,而依魯替尼單藥組僅為45%,無進展生存期也有所改善,風險比達0.559。另一項名為UNITY-CLL的三期臨床試驗(NCT02612311)旨在評估Ublituximab聯合PI3K抑制劑TGR-1202對CLL的安全有效性,臨床試驗以obinutuzumab+苯丁酸氮芥、ublituximab單獨用藥或TGR-1202單獨用藥為參照。該試驗已在進行中,有望在2018年9月達到終點。除了CLL,obinutuzumab聯合TGR-1202治療非霍奇金淋巴瘤的臨床研究也在開展之中。該試驗是一項名為UNITY-NHL的2/3期臨床試驗 (NCT02793583),預期在2019年5月達到終點。除了癌癥外,obinutuzumab也在積極開發多發性硬化的治療方案,兩項隨機雙盲、多中心三期臨床試驗ULTIMATEI(NCT03277261)和ULTIMATEII(NCT03277248)已經開展,用以對比obinutuzumab和特立氟胺在復發性多發性硬化上的安全有效性。這兩項試驗達到終點的預期時間是2021年3月。
處在臨床開發晚期的單抗
除了以上提到的產品外,還有20個單抗產品處在臨床后期,分別是sirukumab,lampalizumab,roledumab,emapalumab,fasinumab,tanezumab,etrolizumab,birtamimab,gantenerumab,anifrolumab,tremelimumab,isatuximab,BCD-100,carotuximab,camrelizumab,IBI308,glembatumumab vedotin,mirvetuximab soravtansine,oportuzumab monatox,L19IL2/L19TNF。其中sirukumab已經在2017年9月遭到FDA拒絕,FDA要求強生補充額外的臨床數據證明sirukumab的安全性,其他19個產品大部分還沒有關鍵性臨床試驗數據,而但這些數據有望在2018下半年或2019年獲得,預期上市時間是2019年至2020年,我們暫且把它們驚喜留到明年介紹。
毫無疑問,單抗的開發正在提速,據估計至2020年獲批的單抗產品總數將超過100個,同時單抗的市場也將一路高歌,2020年單抗藥物的總市場規模有望觸及1300億美元,2018年,單抗們拭目以待!
Lanadelumab (SHP643,DX-2930)是一種人IgG1單抗,可通過抑制血漿激肽釋放酶而阻止緩激肽的生產。Lanadelumab在三期臨床開展的適應癥為遺傳性血管性水腫(HAE),遺傳血管性水腫是一種以身體受到外界侵害后引發組織水腫為特征的罕見病。Lanadelumab已經獲得美國和歐洲授予的HAE孤兒藥資格和FDA HAE突破性療法認定。2017年5月,夏爾報告了一項為期26周的三期臨床HELP試驗(NCT02586805)數據,該試驗就lanadelumab的安全性和阻止 I 型和II型HAE患者急性血管水腫發生率的療效進行了評估。試驗中,49名患者每二周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 150mg或安慰劑。結果顯示,每二周一次給予lanadelumab 300mg,每四周一次給予lanadelumab 300mg和每四周一次給予lanadelumab 150mg的患者,血管水腫發生率相比安慰劑分別下降87%,73%和76%,試驗達到既定的主要終點和次要終點。另一項開放標簽的三期臨床長期安全有效性研究 (NCT02741596)用以評估lanadelumab阻止 I 型和II型HAE患者急性血管水腫發生的效率,治療終點將在2018年2月到達。本品的上市申請將在2018年初提交到FDA和EMA。
Crizanlizumab
Crizanlizumab (SEG101) 是一種人源化單抗,靶點為血小板選擇蛋白,也就是眾所周知的CD62,研究的適應癥為鐮狀細胞疼痛危象 (SCPC)。鐮狀細胞危象臨床表現為慢性溶血性貧血,易感染和再發性疼痛危象以致慢性局部缺血導致器官組織局部損壞。在安慰劑對照的二期SUSTAIN試驗(NCT01895361)中,198名16到65歲的患者,以5mg/kg的劑量,每四周一次給予crizanlizumab,連續治療50周,SCPC年化率中位值相對安慰劑下降45.3%(1.63 vs 2.98),用藥后首次發生SCPC的時間相比安慰劑延長2.9倍(中位時間分別為4.07月和1.38月),第二次發生SCPC的時間相比安慰劑延長2.0倍(中位時間分別為10.32月和5.09月)。此前,crizanlizumab在美國和歐洲均已獲得SCPC孤兒藥認定,如果PK/PD數據與最終的生產工藝生產的產品一致,諾華將在2018年提交BLA。
Ravulizumab
Ravulizumab (ALXN1210) 是一種人源化,以補體5(C5)為靶點的單抗,是eculizumab的下一代,目前處于注冊前期階段。其開展的臨床試驗包括2項與陣發性睡眠性血紅蛋白尿癥(PNH)相關的三期臨床試驗和2項與非典型溶血尿毒綜合征 (aHUS)相關的三期臨床試驗。這2項(PNH)三期臨床試驗NCT02946463 和NCT03056040是基于一項1/2期臨床試驗的結果。未經補體抑制劑治療的患者使用Ravulizumab后血漿乳酸脫氫酶(LDH)水平快速并持續下降,FACIT疲勞量表得分顯著改善。3期臨床試驗NCT02946463旨在評估Ravulizumab相對eculizumab治療未經補體抑制劑治療的成年PNH患者的安全有效性。該試驗中,患者基于體重在第一天給予一個負荷劑量,第15天給予維持劑量( 40至60kg體重:負荷劑量2400mg, 維持劑量每8周3000mg;60至100kg體重: 負荷劑量2700mg, 維持劑量每8周3300mg;體重大于100kg的患者,負荷劑量3000mg, 維持劑量每8周3600mg),而eculizumab則根據說明給藥。預期的臨床終點是26周內血漿乳酸脫氫酶(LDH)達正常水平的情況。試驗招募了246名患者,將在2017年12月達到終點。另一項3期臨床試驗則旨在評估Ravulizumab對使用eculizumab治療6個月以上且病情穩定患者的療效。Ravulizumab組的給藥方案與前一項臨床試驗相同,eculizumab組的患者則在第183天上給予負荷劑量的Ravulizumab,第197天給予維持劑量,主要終點是26周血漿乳酸脫氫酶(LDH)水平的改變情況。試驗招募了197名患者,試驗將在2018年3月結束。亞力兄弟有望在2018年2季度報告這兩項試驗的結果,并提交BLA。此前Ravulizumab已經獲得美國和歐洲PNH的孤兒藥認定。與aHUS相關的單臂三期臨床試驗NCT02949128招募了55名患者,試驗有望在2018年初結束,另一項關于兒童aHUS的三期臨床試驗NCT03131219也在進行中,結束時間是2018年12月。
Eptinezumab
Eptinezumab (ALD403) 是一個IgG1單抗,靶點為降鈣素基因相關肽(CGRP),開發適應癥是偏頭痛。2017年6月,Alder BioPharma宣布三期臨床 PROMISE1 試驗(NCT02559895)達到主要和次要終點。納入這項研究的陣發性偏頭痛患者平均頭痛天數是8.6天/月,試驗中患者分別給予eptinezumab 300mg、100mg、30mg或安慰劑,最終30mg的低劑量組未納入結果統計。結果顯示,1至12周觀察到患者每月頭痛次數顯著下降,300mg組每月頭痛為4.3天,100mg組為3.9天,安慰劑組為3.2天。接近三分之一的患者在第4-12周的每月頭痛天數下降75%以上,平均有五分之一的患者在1-6月內未至少有一個月未發生頭痛。另一項專門針對慢性偏頭痛設計的三期臨床試驗PROMISE2 (NCT02974153) 已招募1050名患者,該試驗將在2018年上半年結束,Alder計劃在2018年2季度提交上市申請。
Risankizumab
Risankizumab (ABBV066, BI655066)是一種以IL-23的p19亞基為靶點的IgG1單抗,開發適應癥為銀屑病。2017年10月,艾伯維宣布以ustekinumab和adalimumab為對照的三期臨床試驗達終點。在ultIMMa-1(NCT02684370)和ultIMMa-2(NCT02684357)試驗中,以ustekinumab和安慰劑為對照評估了risankizumab的有效性。經過16周的治療,兩項試驗中接受risankizumab治療的患者,達PASI90的患者比率均為75%,而接受ustekinumab患者,分別為 42%和48%,安慰劑則分別只有5%和2%。risankizumab治療組sPGA評分達標率分別為88%和84%,ustekinumab治療組分別為63%和62%,安慰劑組僅為8%和5%。在IMMvent研究(NCT02694523)中,患者分別接受risankizumab或adalimumab治療16周,risankizumab組達PASI90的患者比例為72%,阿達木單抗組則只有47%,risankizumab組sPGA達標率為84%,而阿達木單抗只有60%。本品有望在2018年提交BLA,2019年上市。
Satralizumab
Satralizumab (SA237) 是一種人源化IgG2單抗,靶點為白介素-6受體(IL-6R),開發適應癥為視神經脊髓炎或視神經脊髓炎譜系疾病。評估Satralizumab添加基線治療安全有效性的三期臨床試驗NCT02028884還在進行中,主要的治療終點是30個月內的疾病首次復發時間,該試驗已招募了70名患者,預期將于2018年7月結束。另一項安慰劑對照的三期臨床試驗NCT02073279也已經完成招募,主要的治療終點為38個月內疾病首次復發的時間,該試驗已經招募到98名患者,有望在2018年10月結束。此前該產品已經獲得FDA和EMA關于視神經脊髓炎譜系疾病的孤兒藥認定,有望在2018年下半年提交上市申請。
Brolucizumab
Brolucizumab (RTH258) 是一種單鏈可變區片段,靶點為血管表皮細胞生長因子A(VEGF)-A,開發適應癥為新生血管性老年性黃斑變性(nAMD)。2017年6月,諾華宣布三期臨床試驗HAWK (NCT02307682)和HARRIER (NCT02434328)達治療終點,Brolucizumab治療組48周平均最佳矯正視力相對基線改善情況非劣于aflibercept。這兩項試驗納入nAMD患者超過1800人,分別給予brolucizumab 6mg、brolucizumab 3mg(HARRIER 不涉及)或aflibercept 2mg。結果顯示Brolucizumab 6mg在兩項試驗中都以顯著p值到達主要終點和關鍵次要終點;Brolucizumab 3mg也在HAWK中到達這些終點。此外,在HAWK和HARRIER試驗中,12周用藥一次的患者分別有57%和52%堅持用藥到48周。Brolucizumab表現出良好的耐受性,總體眼睛和非眼睛不良反應發生率都與aflibercept相當。諾華有望在2018年完成低劑量規格的PK試驗,BLA有望在2018年下半年提交。
PRO140
PRO140 是一種人源化IgG4單抗,靶點為CC趨化因子受體5(CCR5),可防止HIV病毒進入T細胞,目前開發適應癥為艾滋病。兩項2/3期臨床試驗(NCT02483078和NCT02859961)已經在2017年10月和2017年12月完成。NCT02483078是一項隨機、雙盲、安慰劑對照的臨床試驗,耐藥的HIV感染者在最佳抗病毒藥的背景治療下,添加PRO140或安慰劑治療。而NCT02859961則研究的是抗逆轉錄病毒治療后病情處于穩定期的患者,換用PRO140單獨治療的有效性。2017年10月,CytoDyn與FDA進行會晤,就提交聯合用藥BLA所需的受試人數和患者類型展開商討,FDA要求NCT02483078試驗完成50例患者的研究,BLA要中提交總數300人的安全性評估。目前該產品已經獲得FDA快速審評通道資格,這也就意味著本品可以提交滾動BLA申請。
Sacituzumab govitecan
Sacituzumab govitecan (IMMU-132) 是一種抗體偶聯物,由人源化anti-Trop-2抗體偶聯伊立替康活性代謝物SN38而成,開發適應癥為晚期三陰乳腺癌 (TNBC)。2017年11月,Immunomedics宣布該公司將在2018年一季度提交BLA,此前本品已經獲得FDA三陰乳腺癌突破性療法認定,支持將BLA的數據來自NCT01631552研究。110例三陰乳腺癌患者接受10mg/kg的sacituzumab govitecan治療,客觀緩解率為34%,臨床受益率為46%,Kaplan-Meier(KM)中位有效期和無進展生存期分別為7.6月和5.5月。驗證本品對復發或難治性三陰乳腺癌療效的三期臨床試驗ASCENT(NCT02574455)已經招募到328名患者,入組的患者均為2種以上方案化療后進展或最少一種方案化療并接受輔助治療后12個月內進展的患者。試驗中患者分別接受sacituzumab govitecan或4個預先設定的單藥治療方案化療,該試驗已經在進行中。
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22,CAT- 8015) 是一種分子量為38kDa的重組銅綠假單胞菌外毒素的毒性部分與CD22抗體可變區片段融合的抗體偶聯物。早在2016年2月,Moxetumomab pasudotox就獲得FDA多毛細胞白血病 (HCL)的孤兒藥認定。本品目前在開展一項復發或難治性HCL的多中心、單臂三期臨床試驗(NCT01829711),該試驗已經在2017年五月達到主要終點,阿斯利康計劃在2018年提交BLA。
Cemiplimab
Cemiplimab (REGN2810, SAR439684) 是一種人程序性死亡受體1(PD-1)單抗,開發適應癥為無法手術的或轉移性皮膚鱗狀細胞癌(CSCC),目前該適應癥已經獲得FDA突破性療法認定。一期臨床試驗NCT02383212顯示本品對局部進展性CSCC和轉移性CSCC均有很好的療效,綜合客觀緩解率為46.2%。目前賽諾菲和再生元正在共同開發Cemiplimab,BLA有望在2018年一季度首次提交。另外一項以無法手術的或轉移性CSCC為研究對象,名為EMPOWER-CSCC1的二期臨床試驗也在開展中。除了CSCC,賽諾菲和再生元還在積極開展宮頸癌(NCT03257267)和非小細胞肺癌肺癌(NCT03088540)的三期臨床試驗,試驗終點將分別在2020年5月和2021年11月達到。
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101)是一種糖基化的嵌合型抗體,靶點為CD20。該抗體含低含量的巖藻糖,可改善FcγRIIIa的結合率,抗體依賴的細胞介導的細胞毒性作用(ADCC)相比利妥昔單抗有所增強,開發適應癥為慢性淋巴瘤白血病 (CLL)。一項多中心、開放標簽的三期臨床GENUINE試驗(NCT02301156)評估了ublituximab與依魯替尼聯合用藥相對依魯替尼單獨用藥的安全有效性。該試驗共有126名CLL患者入組,患者每日一次給予依魯替尼420mg或依魯替尼420mg+ublituximab,結果顯示聯合用藥組客觀緩解率為78%,而依魯替尼單藥組僅為45%,無進展生存期也有所改善,風險比達0.559。另一項名為UNITY-CLL的三期臨床試驗(NCT02612311)旨在評估Ublituximab聯合PI3K抑制劑TGR-1202對CLL的安全有效性,臨床試驗以obinutuzumab+苯丁酸氮芥、ublituximab單獨用藥或TGR-1202單獨用藥為參照。該試驗已在進行中,有望在2018年9月達到終點。除了CLL,obinutuzumab聯合TGR-1202治療非霍奇金淋巴瘤的臨床研究也在開展之中。該試驗是一項名為UNITY-NHL的2/3期臨床試驗 (NCT02793583),預期在2019年5月達到終點。除了癌癥外,obinutuzumab也在積極開發多發性硬化的治療方案,兩項隨機雙盲、多中心三期臨床試驗ULTIMATEI(NCT03277261)和ULTIMATEII(NCT03277248)已經開展,用以對比obinutuzumab和特立氟胺在復發性多發性硬化上的安全有效性。這兩項試驗達到終點的預期時間是2021年3月。
處在臨床開發晚期的單抗
除了以上提到的產品外,還有20個單抗產品處在臨床后期,分別是sirukumab,lampalizumab,roledumab,emapalumab,fasinumab,tanezumab,etrolizumab,birtamimab,gantenerumab,anifrolumab,tremelimumab,isatuximab,BCD-100,carotuximab,camrelizumab,IBI308,glembatumumab vedotin,mirvetuximab soravtansine,oportuzumab monatox,L19IL2/L19TNF。其中sirukumab已經在2017年9月遭到FDA拒絕,FDA要求強生補充額外的臨床數據證明sirukumab的安全性,其他19個產品大部分還沒有關鍵性臨床試驗數據,而但這些數據有望在2018下半年或2019年獲得,預期上市時間是2019年至2020年,我們暫且把它們驚喜留到明年介紹。
毫無疑問,單抗的開發正在提速,據估計至2020年獲批的單抗產品總數將超過100個,同時單抗的市場也將一路高歌,2020年單抗藥物的總市場規模有望觸及1300億美元,2018年,單抗們拭目以待!
處在臨床末期的單抗藥物

聲明:本文的內容大量參考了Antibodies to watch in 2018一文,但本文不是翻譯文章,筆者在原文的基礎上進行漢化,信息進一步補充或刪減,加入了市場相關的內容和作者的觀點,因此本文是唯一的。此外,藥事縱橫刊發本文僅用于學習和交流,轉載此文務必注明作者和來源,否則一律視為惡意侵權,侵權必究。對于文章觀點產生異議的朋友,可參考英文原文。地址:
參考文獻:Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/

參考文獻:Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/
?版權所有 重慶派金生物科技有限公司? 渝ICP備18013524號-1

聯系方式
15823078768 劉先生??
??地址:重慶市北碚區豐和路106號??
pegbio@pegbiocq.com
搜索
